Article Text

Abstract
A 59-year-old white man with known myeloproliferative neoplasm (MPN) and myelodysplastic syndrome (MDS) presented with worsening leucocytosis and thrombocytosis in the setting of a presumed infection. The patient had been diagnosed 2 years earlier with an MPN/MDS overlap syndrome, based on characteristic mutations in JAK2, IDH1 and SRSF2. During his current evaluation, he was noted to have new microcytosis, with a mean corpuscular volume of ~70 fL down from his baseline of ~90 fL. His laboratory workup showed normal iron studies, normal haemoglobin electrophoresis, and no evidence of haemoglobin H or mutations in his ATRX coding region. Without any identifiable cause of his new microcytosis, he was given a presumptive diagnosis of acquired thalassemia in the setting of his unusual MPN/MDS overlap syndrome.
- haematology (incl blood transfusion)
- medical education
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Here, we present a patient with acquired microcytosis developing out of the background of an unusual combined myeloproliferative/myelodysplastic overlap syndrome. This case raises awareness of the rare entity of acquired microcytosis, also known as acquired thalassemia, particularly in patients with underlying haematological disorders.
Case presentation
Approximately 2 years prior to presentation (figure 1, grey arrow), this man underwent evaluation for an abnormal peripheral blood smear that demonstrated the presence of immature white blood cells concerning for myeloid blasts. A bone marrow biopsy evaluation demonstrated a hypercellular marrow with evidence of myeloid dysplasia. There were no ringed sideroblasts. Mutational analysis done at the time confirmed the presence of mutations in three genes: JAK2, IDH1 and SRSF2 (table 1). While the JAK2V617F mutation is diagnostic of a myeloproliferative neoplasm (MPN), the IDH1 and SRSF2 mutations are commonly associated with myelodysplastic syndromes (MDS).
{kind=link}
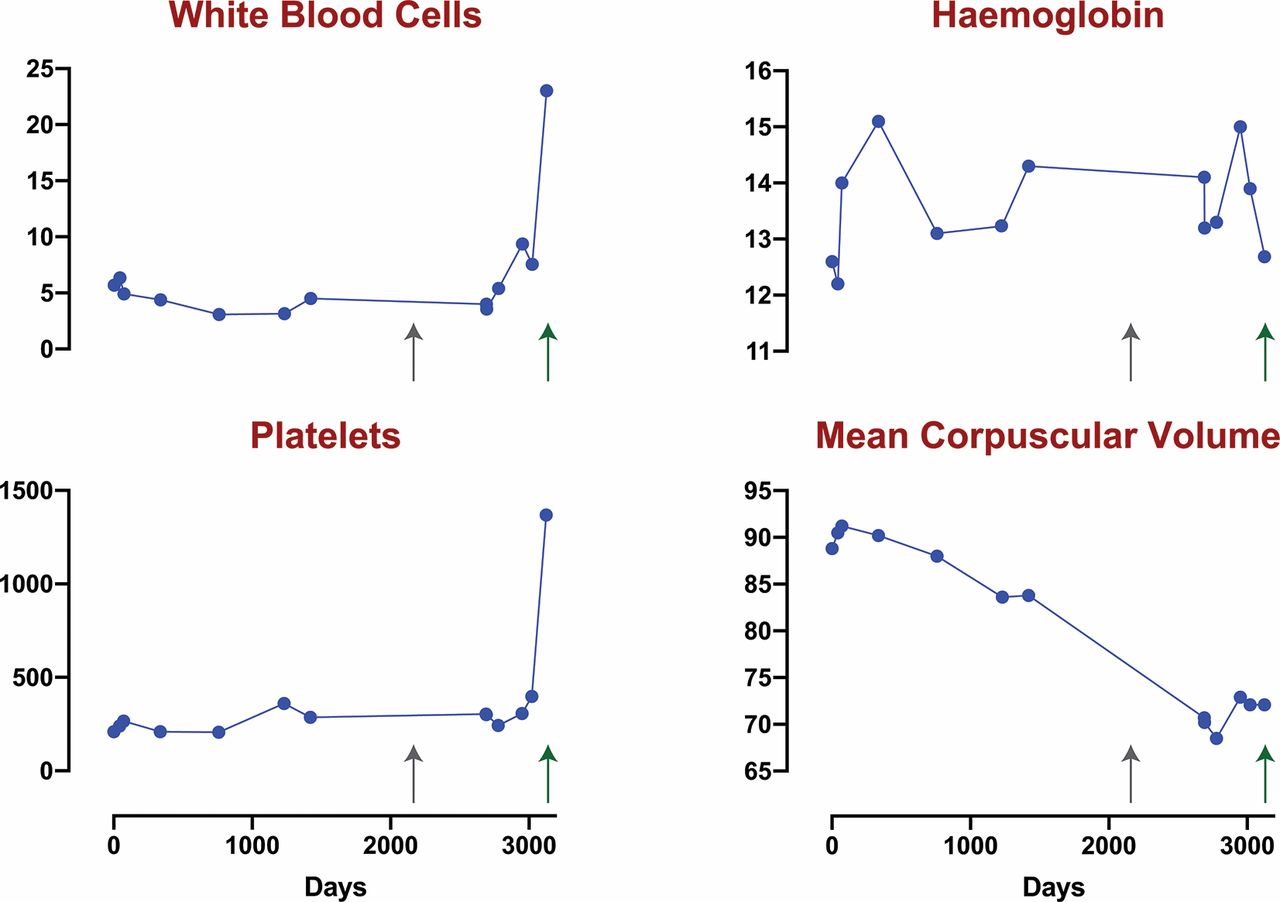
A timeline showing the progression of elements of the patient’s complete blood count. HGB, haemoglobin; MCV, mean corpuscular volume; PLT, platelets; WBC, white blood cell.
Spectrum of mutations detected on targeted gene sequencing analysis of the bone marrow
He had a history that was notable for amyotrophic lateral sclerosis (ALS), coronary artery disease and prostate cancer treated by radical prostatectomy. His ALS had progressed to the point where he could only communicate using his eyes to move the cursor on a keyboard associated with a voice generator.
He was admitted to the hospital for unintentional weight loss and worsening leucocytosis and thrombocytosis.
On admission (figure 1, green arrow), his laboratory evaluation revealed a white blood cell count of 29 000/μL, a haemoglobin of 119 g/L and a platelet count of 1 629 000/μL. His mean corpuscular volume (MCV) was 72.3 fL, down from his baseline of ~90 fL. His complete blood count laboratory values over the last 3 years are shown (figure 1).
At its nadir, his MCV was 71.6 fL with an associated red blood cell count of 6.1 (million cells/μL), resulting in a Mentzer index of 11.7.1 Iron studies demonstrated a ferritin of 520 μg/L (20–300), total iron binding content 206 μg/dL (230–400) and iron 47 μg/dL (45–160). His serum vitamin B12 and serum folate were normal. Serum copper and serum zinc levels were normal. His comprehensive metabolic panel was normal, including total and indirect bilirubin. Lactate dehydrogenase was 511 U/L. The erythrocyte sedimentation rate was 13 mm/hour (0–13). Abdominal imaging revealed splenomegaly—which would be expected in the setting of his MPN—and no other abnormalities.
The peripheral blood smear confirmed his microcytosis. Haemoglobin electrophoresis showed a very mildly elevated haemoglobin A2 but was otherwise normal. Gene sequencing of his haemoglobin alpha chains did not reveal any abnormalities. The mutational analysis was repeated and revealed the presence of an additional mutation in the ASXL1 gene that was not detected during his initial profiling (table 1). Of note, no mutation in the ATRX coding sequence was identified.
The patient was treated for his presumed infection and discharged home after 6 days.
Outcome and follow-up
The patient was discharged from the hospital after his infection clinically improved. While his haematological problems remained fairly stable, the patient passed away due to complications of his ALS a few months after being discharged.
Discussion
In general, microcytosis results from conditions that lead to a disruption of alpha or beta-globin-chain production. Most commonly, microcytosis is the result of decreased iron availability or an underlying inherited haemoglobinopathy such as thalassemia. The Mentzer index is one way of quickly discerning between thalassemia and iron-deficiency, with an MCV/RBC ratio <13 suggesting thalassemia.1 In our patient, the value of 11.7 in conjunction with his iron studies was consistent with thalassemia.
Decreased iron availability may be due to an absolute lack of iron stores (ie, iron deficiency), or to a functional lack of available iron. A functional iron deficiency can manifest in the setting of a chronic inflammatory state where serum hepcidin levels are elevated or in the setting of sideroblastic anaemia.2 In this case, the patient’s other comorbidities may have precipitated some chronic inflammation, though his erythrocyte sedimentation rate of only 13 mm/hour argues against an overwhelming inflammatory state.
The exact mechanism by which red blood cells become microcytic is not well documented. The predominant theory is that red blood cells stop dividing once they reach a certain concentration of intracellular haemoglobin. As the production of haemoglobin slows down, more divisions occur before this concentration is reached, resulting in smaller cells.3 For this reason, microcytosis serves as a loose indicator of decreased haemoglobin synthesis, and macrocytosis an indicator of decreased DNA production.
Diagnosing the aforementioned conditions is fairly standard, with evaluation of the peripheral blood smear, a complete CBC, reticulocyte count, iron studies, vitamin B12 and folate stores, and erythrocyte sedimentation rate. A bone marrow biopsy is required for identification of ringed sideroblasts in clinching the diagnosis of a sideroblastic anaemia.2 The haemoglobin electrophoresis is useful in cases of beta-thalassemia and certain cases of alpha-thalassemia (where 3 or 4 copies of the alpha-globin gene are defective resulting in the production of HbH). Alpha-globin and/or beta-globin gene sequencing can also be undertaken, though this is most often done as part of family planning when both parents are suspected of being carriers of a silent mutation.4
Much less common are cases of acquired microcytosis or acquired thalassemia. Here, patients have had to acquire a haematopoietic stem cell germ-line mutation in a one of the globin loci or in a regulatory region or gene controlling globin-chain production.
MDS is most typically associated with an acquired macrocytosis.5 Acquired microcytosis occurs in a very small subset of patients with known MDS or other haematological disorders such as primary myelofibrosis, acute myeloid leukaemia and essential thrombocytosis.6 While alpha-thalassemia is more common, beta-thalassemia associated with haematological disease has also been reported.7
Mutations in the non-globin gene ATRX have been demonstrated to cause acquired thalassemia in patients with MDS.7 ATRX is a chromatin remodelling gene that appears to play a role in α-globin chain production. Patients with inherited mutations of ATRX show alpha-thalassemia as well as developmental delay, the pathology after which the gene is named. When an acquired ATRX mutations occurs in the haematopoietic stem cells, alpha-globin chain expression is reduced without affecting the protein structure.8
The phenomenon has recently been designated alpha-thalassemia associated myelodysplastic syndrome (ATMDS), and a registry database was established for the collection of patient data. Over 100 different missense mutations have been registered, and new cases are still being reported.9–12 Diagnosis of ATMDS depends on the detection of HbH by haemoglobin electrophoresis or examination of the peripheral blood smear and has only been seen in the context of another haematological disorder.7 MDS appears to be the most commonly associated condition, and 43% of patients with MDS and new microcytosis were found to have mutations in ATRX.11
While ATMDS is a well-documented cause of thalassemia in patients with MDS, MDS/MPN overlap syndrome has no such strong association to date. The diagnosis of MDS/MPN overlap depends on genetic profiling, and MDS and MPN/MDS overlap share some common mutations. JAK2 is one of the most common mutations found in MPN, but it is found in a small minority of MDS patients.12 ASXL1 is more common in MPN and MDS/MPN overlap patients, and the most common mutation in MDS/MPN overlap, with a male predominance.13 14 This patient’s mutation burden in IDH1 is atypical for MDS/MPN overlap syndrome.14
When evaluating for the cause of microcytosis, a longitudinal evaluation can be very helpful. The timing and rate of decline of the MCV can be informative in determining the aetiology. Our patient’s microcytosis appears to have developed over the course of almost 4 years. His decline in MCV was slow but consistent, beginning at an MCV of roughly 90 and decreasing to a nadir of 68.5. This very gradual decline in MCV suggests a more insidious and chronic process than an iron deficiency or anaemia of chronic disease. It remains unclear whether the timeline of the development of microcytosis is connected with progression in mutation burden.
Learning points
Microcytosis has several common causes, but an unremarkable initial workup may warrant a more thorough investigation.
Acquired microcytosis has been associated with certain haematological disorders, most commonly myelodysplastic syndromes and most commonly affecting production of alpha-haemoglobin.
Elements of the timeline and severity of microcytosis can offer clues to its aetiology.
Microcytosis is caused by increased nucleic acid synthesis and progenitor divisions relative to haemoglobin production.
Footnotes
Contributors MM wrote the manuscript with support from TK and DBS.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.