Article Text

Abstract
Antemortem assessment of sporadic Creutzfeldt-Jakob disease (sCJD) can be significantly hampered due to its rarity, low index of clinical suspicion and its non-specific clinical features. We present an atypical case of definitive sCJD. The patient died within 5 weeks of the disease onset. This unusually short duration of disease presented a significant diagnostic dilemma. The patient presented with 2-week history of sudden-onset cognitive decline, memory loss, aphasia and ataxia. MRI Diffusion-weighted sequences revealed cortical ribboning sign without cerebral atrophy. Protein 14-3-3 from cerebrospinal fluid (CSF) was detected, and postmortem brain autopsy confirmed the diagnosis of sCJD. This case underscores the importance of considering CJD as a potential diagnosis for rapidly progressive dementia. Serology tests, EEG, MRI and CSF study are invaluable diagnostic tools when assessing for sCJD. Appropriate use of those diagnostic tests, along with a detailed clinical examination, can successfully and promptly exclude other differential diagnoses and confirm sCJD.
- Neurology
- Infection (neurology)
- Memory Disorders
- Neuroimaging
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Creutzfeldt-Jakob disease (CJD) is an invariably fatal but rare infectious spongiform encephalopathy secondary to infectious prion protein. The annual incidence rate is known to be approximately 1–2 per million worldwide.1 CJD is classified according to its aetiology into four subtypes of sporadic, familial, iatrogenic and variant forms.2 Sporadic CJD (sCJD) occurs when normal prion protein PrPc spontaneously convert into infectious form PrPSc through an unknown process. It consists of up to 85%–90% of all the CJD cases, and its mean age of onset is known to be at 65 year.2 3 Most patients will pass away due to infection or cardiorespiratory failure within 12 months of disease onset, and both genders seem to be equally affected.1 4 Familial CJD is autosomal dominant, and this form is secondary to the mutation of the prion protein gene (PRNP, also known as CD230) on the short arm of chromosome 20.2 5 It consists of 5%–10% of the total CJD cases and is known to be correlated with a relatively early onset of age (40–50 years). Its disease duration can be up to 24 months, which is relatively longer than those of other subtypes.6 Variant CJD (<1%), which is reported since the middle of the 90s and associated with ingestion of meat products infected with prion, leads to cerebral prion plaques rather than spongiform alteration in the brain.7 8 Iatrogenic CJD (<1%) is an acquired form of CJD secondary to transmission from an established source; previous human pituitary hormone injection, use of contaminated neurosurgical instruments, dural graft transplant or corneal transplant have been attributed.2 8 sCJD can present with rapid cognitive and functional decline, memory deficit, myoclonus, pyramidal/extrapyramidal signs and visual deficits.2 These typical clinical features are well summarised in the WHO and MRI-CJD Consortium diagnostic criteria, which provide an invaluable diagnostic guideline to any physicians unfamiliar with the disease.2 9 However, even those clinical signs mentioned on the guidelines are non-specific and can also manifest in other neurological differential diagnoses of rapidly progressive dementia. Given the rare incidence and non-specific clinical features of CJD, it is not uncommon for it to be missed as a potential diagnosis. Furthermore, sCJD can present with atypical clinical features outside the guidelines, thereby posing a significant diagnostic dilemma.10 Hence, in addition to a detailed clinical history and examination, early use of appropriate investigations involving MRI, EEG, blood and cerebrospinal fluid (CSF) analysis is crucial in excluding other differentials and at the same time, confirming probable sCJD. Here, we discuss a highly unusual case of a rapidly progressing sCJD characterised by an extremely short duration of disease of <5 weeks. Despite the initial presenting clinical features of rapid cognitive decline, aphasia, ataxia and visual disturbances, which are typical of sCJD, the patient was initially worked up for stroke given the acuteness of his disease onset. Nevertheless, additional investigations with MRI, EEG and CSF study allowed for prompt diagnosis of probable CJD, with postmortem brain autopsy confirming definitive sCJD.
Case presentation
The patient was a 76-year-old man who was referred to emergency department (ED) by his general practitioner (GP) for investigation of suspected stroke after a 2-week history of sudden-onset motor, functional and cognitive decline characterised by confusion, memory loss, visual deficit and gait unsteadiness. According to the family, he had migrated to Australia from Thailand 30 years ago, and he was fluent in both Thai and English. He was both physically and cognitively well prior to the onset of symptoms, and he was not known to have any medical or psychiatric records, previous surgeries, blood transfusion or hormonal injection. He was not on any regular medications. No family history of dementia or prion disease was reported. Two weeks before his presentation, he was reviewed by his GP and was initially commenced on an oral antibiotic for delirium secondary to suspected urinary tract infection. However, when there was no improvement, the patient was referred to ED.
On initial assessment at ED, he appeared confused. He demonstrated loss of ability to speak or read English, though he did not display visual agnosia when prompted in Thai (translated by his family). He exhibited severe slurring of speech, dysgraphia and ataxic gait. There was no facial asymmetry. Extraocular movements were intact. He was slow to obeying commands and demonstrated loss of ability to perform complex tasks such as drawing a full clock, though he was able to perform one-command tasks when prompted in Thai. Full cranial nerve examination revealed left-sided visual neglect. He had grossly normal tone, power, sensation and reflexes. Babinski reflexes were absent. He could only mobilise with one assist on seat-walker due to ataxic gait. No myoclonic jerks were identified.
The patient underwent markedly rapid cognitive decline, and by day 7 since admission, episodes of severe agitations, akinetic mutism, aphasia, echolalia and echopraxia, and stiffness of limbs with upper limb myoclonic jerks were observed. Rowland Universal Dementia Assessment Scale (RUDAS) test at this time scored 0, with many of the items unable to be assessed. He also demonstrated a rapid functional decline to the point of being bedbound and incontinent of bowel and urine. He developed loss of swallowing function, therefore requiring nasogastric (NG) feeding.
Investigations
Initial CT brain did not show any intracranial pathology (figure 1). Basic full blood count, blood sugar level, liver and renal panels, electrolytes and vitamin B12 level were normal. Thyroid function test (including anti-thyroid peroxidase), which was performed to rule out Hashimoto encephalopathy, came back as normal. Inflammatory markers (C reactive protein and erythrocyte sedimentation rate) were normal and autoimmune screen (Antinuclear Antibody (ANA), Extractable nuclear antigen (ENA), Anti-neutrophil cytoplasmic antibodies (ANCAs), anti-double stranded DNA (dsDNA)), HIV, serum VDRL, serum cryptococcal antigen, paraneoplastic panel (including anti-Hu, anti-Jo) and heavy metal screening were negative.

CT brain (non-contrast) on initial presentation to emergency department. There is no infarct, haemorrhage, mass or focal intracranial abnormality. There are no features of normal pressure hydrocephalus.
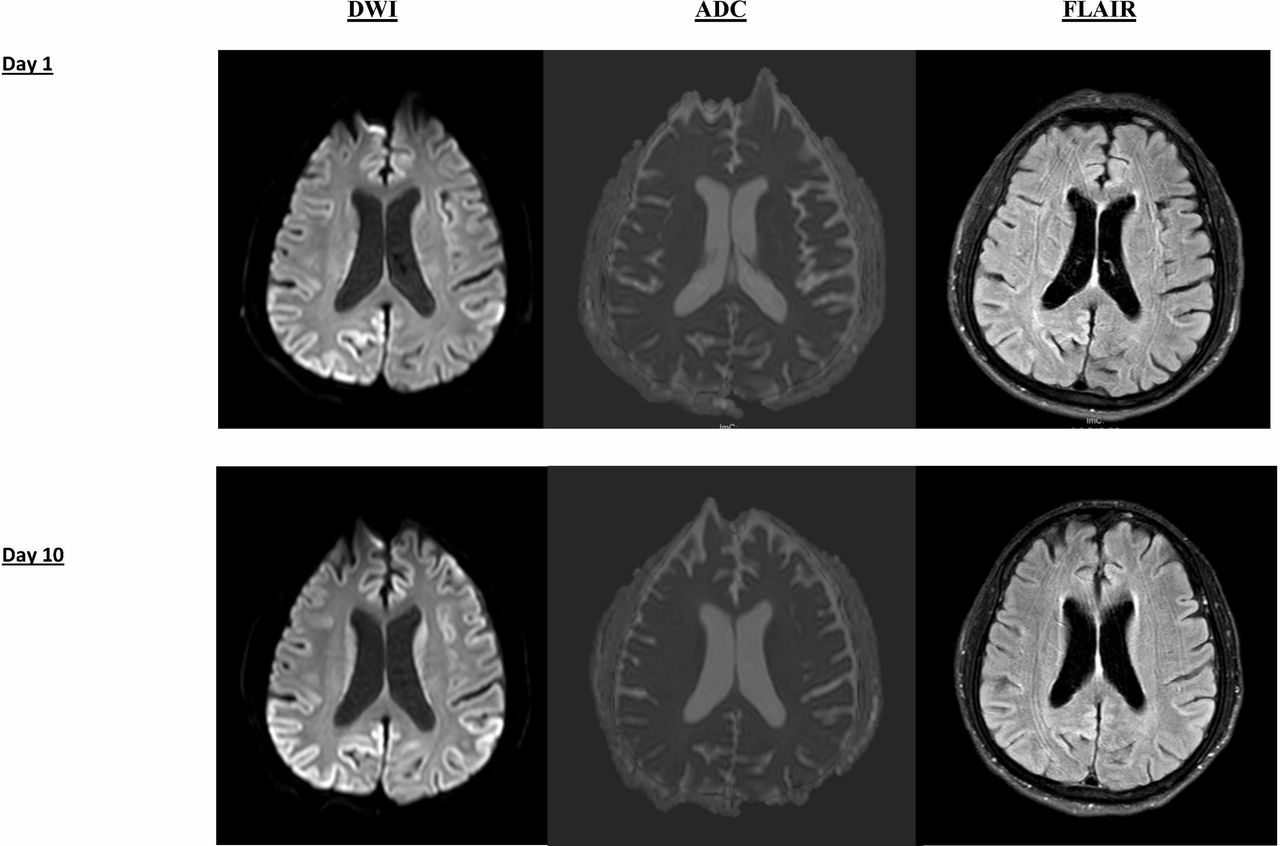
MRI brain with gadolinium on the day after his admission (figure 2) revealed no stroke but bilateral cortical ribboning on diffusion-weighted images (DWI) and fluid-attenuated inversion recovery (FLAIR), suggestive of sCJD. Following the MRI result, lumbar puncture was performed to investigate for CJD via 14-3-3 CSF assay at the reference laboratory (Florey Medical Institute, Melbourne). Video EEG on day 6 postadmission revealed an abnormal EEG pattern suggestive of diffuse encephalopathy, which was characterised by posterior dominant rhythm in 5–7 Hz range with frequent runs of delta activity (1–2 Hz) and occasional runs of diffuse sharply contoured theta activity. No triphasic, periodic wave complexes or epileptiform discharges were recorded.

{kind=link}
{kind=link}
MRI brain on day 1 and day 10 of hospital admission. DWI and FLAIR sequences are shown. DWI demonstrates cortical ribboning (signal hyperintensity of cerebral cortical gyri) of the regions. The corresponding FLAIR sequence shows less-intense cortical ribbon sign, however. Cortical ribboning is highly suggestive of sporadic CJD. There was no significant interval change in size or pattern of distribution from day 1 to day 10 hospital admission. No signal hyperintensity was found in putamen or caudate nucleus. CJD, Creutzfeldt-Jakob disease; DWI, diffusion-weighted imaging; FLAIR, fluid-attenuated inversion recovery
Repeat MRI on day 10 (figure 2) revealed no significant interval change in the pattern and distribution of cortical ribboning despite severely marked clinical deterioration observed. Result for the CSF assay for 14-3-3 came back as positive. No cells were seen in CSF. Glucose and protein were within the normal range. No oligoclonal band was detected. Other CSF tests for bacterial, fungal, tuberculosis, cryptococcal antigen, antimeasle antibody, viral encephalitis (Herpes Simplex virus, Varicella-Zoster virus, Epstein Barr virus, and Cytomegalovirus), syphilis and heavy metal screening were negative.
His clinical signs, as well as his MRI findings and CSF finding fulfilled the diagnostic criteria for probable CJD as per MRI-CJD Consortium Criteria.6 11
Differential diagnosis
A number of differential diagnoses were considered and evaluated through various investigations. Table 1 shows investigations performed and differential diagnoses ruled out based on the investigation results.
Investigations performed to rule out differential diagnoses of rapidly progressive dementia
Treatment
While waiting for the CSF result, he was empirically treated with intravenous acyclovir for viral encephalitis, which did not result in any improvement. An empirical trial of 5 days of intravenous methylprednisolone commenced on day 7 for autoimmune encephalitis showed no clinical improvement either. He was treated with quetiapine for agitation and valproate for myoclonic jerks with minimal effect.
Outcome and follow-up
By day 14 postadmission, NG feeding was ceased due to the patient repeatedly pulling it away. He was referred to palliative care, and he passed away on day 17. Postmortem brain autopsy was positive for CJD, with sections of neocortex showing widespread microvacuolar spongiform change, most prominent in the occipital lobe and superior temporal lobe sections with associated gliosis and mild neuronal loss. Other sections of the brain, including striatum, thalamus and cerebellum also showed mild to moderate degree of microvacuolar spongiform change. Immunohistochemistry for 12F10 demonstrated extensive staining in the frontal lobe, basal ganglia and cerebellum. No florid plaques were identified.
Discussion
CJD is a rare and untreatable disease, which has an annual incidence of 1–2 per million worldwide.2 9 CJD can be divided into sporadic, familial, iatrogenic and variant forms.6 In our case, the patient had sCJD.
Our case demonstrated the importance of early consideration of CJD as a potential diagnosis for rapidly progressive dementia. The case was diagnosed, as per WHO and MRI-CJD Consortium diagnostic criteria, as a probable sCJD during the antemortem stage based on the clinical findings, positive MRI and CSF analysis. Diagnosis of CJD can be challenging. Its common clinical features are non-specific and can be present in other differential neurodegenerative diseases. Atypical clinical features not fulfilling diagnostic criteria and the absence of any significant findings in EEG and MRI may further hamper a diagnosis of CJD. Therefore, it is not uncommon for CJD to be missed or diagnosed late, and its clinical presentations can be wrongly attributed to other more common differential diagnoses. In other cases, physicians may even request for highly invasive antemortem brain biopsy as they may be unsure of readily available, non-invasive diagnostic tests.
As summarised on the criteria, common clinical findings of sCJD include rapid cognitive and functional decline, memory deficit, personality change, pyramidal/extrapyramidal and cerebellar signs, visual deficits and myoclonus.1 2 Akinetic mutism may be observed in the later stage of the disease.1 In our case, the patient had presented with many of the aforementioned typical clinical features. Furthermore, there was no relevant family history or previous surgical history (including the hormonal injection or blood transfusion), thereby ruling out the possibility of familial or iatrogenic forms.
Brain MRI is one of the most informative diagnostic tools when assessing sCJD antemortem. Cortical ribbon sign (hyperintensity on the cortical gyri on the DWI and FLAIR sequences), which was detected in the case, is one of the known characteristic features of CJD.6 MRI can also assist with excluding other subtypes of CJD, such as the variant subtype of CJD. sCJD is typically demonstrated by cerebral atrophy or increase in signal intensity (especially on a DWI sequence) in the cerebral cortex, putamen and caudate nucleus.6 11 As for other forms of CJD, pulvinar sign, basal ganglia and cortical hyperintensity are predominantly found in vCJD and fCJD, respectively.2 MRI can detect the characteristic sCJD changes reliably according to recent studies, with 91% sensitivity and 95% specificity with DWI and FLAIR sequences.6 Moreover, studies have also shown that sensitivity was superior for DWI than FLAIR.6 12 Therefore, MRI, as a non-invasive investigation, should be performed for any patients with clinical features of CJD.
EEG is another important investigation when evaluating a patient with potential CJD. Periodic sharp-wave complexes (PSWC) have been demonstrated in up to two thirds of patients of sCJD. PSWC typically occur as one per second periodic triphasic complex in the temporal region of the brains.2 This distribution indicates the prodromal stage of disease onset. As the disease progresses, the disease will manifest with a global bifrontal distribution of PSWC. However, the absence of PSWC on EEG does not exclude a diagnosis of CJD. Patients with CJD can also exhibit diffuse, non-specific slow EEG background patterns, which may be observed in encephalopathy of various neurodegenerative diseases other than CJD. Moreover, PSWCs are known to be correlated with MV1 and MM1 genotypes.2 13 In our patient, non-specific runs of delta activity of around 1–2 Hz as well as diffuse sharply contoured theta activity were seen. However, there were no triphasic, periodic wave complexes or epileptiform discharges. Although typical EEG findings may not be observed in the terminal stage of the disease, this investigation is reported to have high specificity of 100% but low sensitivity of 37.5%.14
Another invaluable diagnostic test is the CSF analysis. This investigation is recommended invariably in patients with rapidly progressive dementia to exclude other differential neurodegenerative diseases. It is also an invaluable test for CJD. The CSF study for CJD is tested at certain national reference centres with expertise on CJD. In our case, the CSF sample was sent to Florey Institute of Neuroscience in Melbourne to detect 14-3-3 protein, which is a surrogate marker of neuronal destruction released into CSF. CSF protein levels rarely rise in CJD but rise in this particular marker can indicate probable CJD. As 14-3-3 protein can be also detected in other non-prion diseases, including viral encephalitis, stroke or Hashimoto encephalitis, the test result must be interpreted in the context of clinical findings. If 14-3-3 protein assay is performed in an appropriate clinical setting, then the test can demonstrate high sensitivity and specificity for the disease.13 Near-future analysis of CSF for CJD will likely involve prion protein conversion assay, known as the real-time quaking-induced conversion (RT-QuIC), which has been developed recently. This test can provide a definitive diagnosis of CJD from CSF sample by detecting PrPScsc (infectious form of prion). The test is known to have 80%–90% sensitivity and up to 100% specificity.15 Although this test is not yet part of the diagnostic criteria for sCJD, this novel test has a great potential to increase accuracy of antemortem diagnosis of sCJD.2 15
The gold-standard investigation for the diagnosis of CJD remains to be a brain biopsy. Despite being a definitive test, the frequency of a positive biopsy result is low with 54%.16 Another study also found that 42% of the biopsies were non-diagnostic.14 The main reasons for the low success rate include: first, the samples must show vacuolation when investigated histopathologically, but this may be missed. Second, the biopsy samples must contain a sufficient level of amyloid deposits to be positive in an immunohistochemical test for PrP, and not all of the brain regions can be examined for histological changes.2 Moreover, the test itself is highly expensive and is rarely indicated; it is performed only if the case is highly suspicious of CJD when other non-invasive tests are negative or at the request of the patients or family members. Therefore, a diagnosis of CJD should depend on a combination of clinical findings and non-invasive investigations including serology, MRI, EEG and CSF analysis. In our patient, a postmortem brain autopsy was performed with family consent. The sample revealed the typical spongiform alteration, gliosis and neuronal loss with detection of prion protein via immunochemistry, thus confirming the diagnosis of definitive sCJD.
Learning points
Known diagnostic criteria for Creutzfeldt-Jakob disease (CJD) must be used in conjunction with a detailed history and appropriate investigations when evaluating patients presenting with rapidly progressive dementia.
CJD can pose a significant diagnostic challenge when accompanied by non-specific or atypical features which differ from known diagnostic criteria.
CJD must be considered as a potential diagnosis for patients who present with rapidly progressive dementia without any evidence of infection or stroke.
A high index of clinical suspicion for CJD, exclusion of differential diagnoses of cognitive decline and appropriate use of available diagnostic tools, including MRI, cerebrospinal fluid analysis and EEG, are crucial in accurately and promptly evaluating a patient with suspected CJD.
MRI brain with diffusion-weighted imaging and FLAIR sequences should be performed for all patients with suspected CJD. However, MRI findings do not appear to provide information on clinical severity or prognosis.
References
Footnotes
Contributors GTK: took history, examined the patient, collected data (including images) and wrote up the abstract, background, case presentation sections as well as the discussion sections. MSK: wrote up the discussion section of the case report and edited the case report.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Next of kin consent obtained.