Article Text

Abstract
A 72-year-old woman presented with nephrotic proteinuria and moderate haematuria. Renal pathology was compatible with immunotactoid glomerulopathy (ITG), for which there is no consensus for appropriate therapy. We, therefore, postponed immunosuppressive therapy. After 4 years, the patient’s renal function started to decline and renal pathology was re-evaluated, revealing a pathological change from mesangial proliferative glomerulonephritis to endocapillary proliferative glomerulonephritis. Treatment with oral prednisolone (30 mg/day) was initiated. Within 5 weeks, complete remission of proteinuria was obtained (proteinuria 6.02 g/gCr to 0.12 g/gCr), and the patient’s renal function stabilised. Generally, responsiveness to immunosuppressive therapy is poor in patients with ITG, and the present case represented a very rare clinical course. Some previous cases have indicated susceptibility to the therapy, regardless of the severity of renal damage. As a possible distinct entity that determines susceptibility to immunosuppressive therapy, we suggest the presence of a latent lymphoproliferative disease with no significant haematological symptoms.
- nephrotic syndrome
- proteinurea
- pathology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Immunotactoid glomerulopathy (ITG) is a rare disease characterised by organised deposits composed of parallelly arranged microtubules in the glomerulus. ITG is a progressive disease that can lead to end-stage renal disease (ESRD) in the short term. However, the clinical course is difficult to predict because few series have been published, and there is no consensus for the appropriate therapy. Responsiveness to immunosuppressive therapy is generally poor in ITG, with a reported rate of partial response of <10%.1 2 Several pathological forms of ITG have been reported previously, but it is not yet clear whether each has a different aetiological background or differs only on the basis of time course.
Case presentation
A 72-year-old woman with a history of hypertension, type 2 diabetes, dyslipidaemia and brain infarction was referred to our hospital for evaluation of proteinuria. The patient had taken a hypoglycaemic agent since the age of 50, and her glycaemic state was generally stable with this medication. Haemoglobin A1c was 6.0% and diabetic retinopathy was not observed. Her other medications included a statin, angiotensin Ⅱ receptor blocker, calcium-channel blocker, furosemide and aspirin. On admission, physical examination revealed no notable findings and her blood pressure was generally controlled under 130/80 mmHg.
Investigations
Until the age of 71, no proteinuria had been reported for the patient and urinary occult blood was (−)–(±). In October 2011, at the age of 71, urinalysis revealed proteinuria of 1+, which had subsequently increased gradually. On admission, urinalysis revealed proteinuria of 2+,red blood cells of 20–29/high-power field, and no casts, while 24 hours urine collection revealed proteinuria of 1.28 g/day. Other relevant laboratory data included total protein of 6.6 g/dL, albumin of 4.3 g/dL, creatinine 0.68 mg/dL and estimated glomerular filtration rate (eGFR) 64 mL/min. Lipids were within normal reference ranges. Although serum C3 was slightly low (66 mg/dL), serum immunoglobulins (IgG, IgA, IgM) and C4 levels were within normal reference ranges. We did not detect anti-DNA, anti-glomerular basement membrane or anti-neutrophil cytoplasmic antibodies, or any antibodies against hepatitis B, hepatitis C or HIV. Cryoglobulins were not detected, and protein immunoelectrophoresis of serum and urine did not show paraprotein. Abdominal sonography revealed normal-sized kidneys.
In May 2013, at the age of 73, the patient underwent a renal biopsy. Under light microscopy, a total of 12 glomeruli were observed; one glomerulus exhibited global sclerosis. The mesangial matrix and cells were diffusely increased to a mild extent. Mesangial interposition was not observed. Mild interstitial fibrosis and tubular atrophy were focally observed, as was mild interstitial infiltration of lymphocytes (figure 1A,B). Immunofluorescence studies could not be performed because no glomerulus was contained in the specimen. Under electron microscopy, dense deposits were observed in the mesangial matrix to a moderate extent. These deposits consisted of microtubular structures that had hollow cores and were arranged in parallel arrays with an average diameter of 40 nm at a higher magnification. No subendothelial, epithelial space or basement membrane deposits were observed (figure 1C). We thus diagnosed the patient as having ITG. Patients with this disorder are reported to have a relatively high incidence of lymphoproliferative disease,3 but further laboratory testing and a computed tomographic scan failed to reveal findings of lymphoproliferative disorders. Since the patient’s renal function and proteinuria were stable after the first biopsy, the existing antihypertensive therapy was continued.

(A,B) Light microscopic findings from the first biopsy showed diffusely increased mesangial matrix and cells to a mild extent without mesangial interposition. (A) Periodic acid-Schiff. 400× and (B) Periodic acid-Schiff-methenamine silver. 400×. (C) Electron microscopic findings showed dense deposits in the mesangial matrix, which had hollow cores and were arranged in parallel arrays with an average diameter of 40 nm at a higher magnification (4000×, 40 000×).
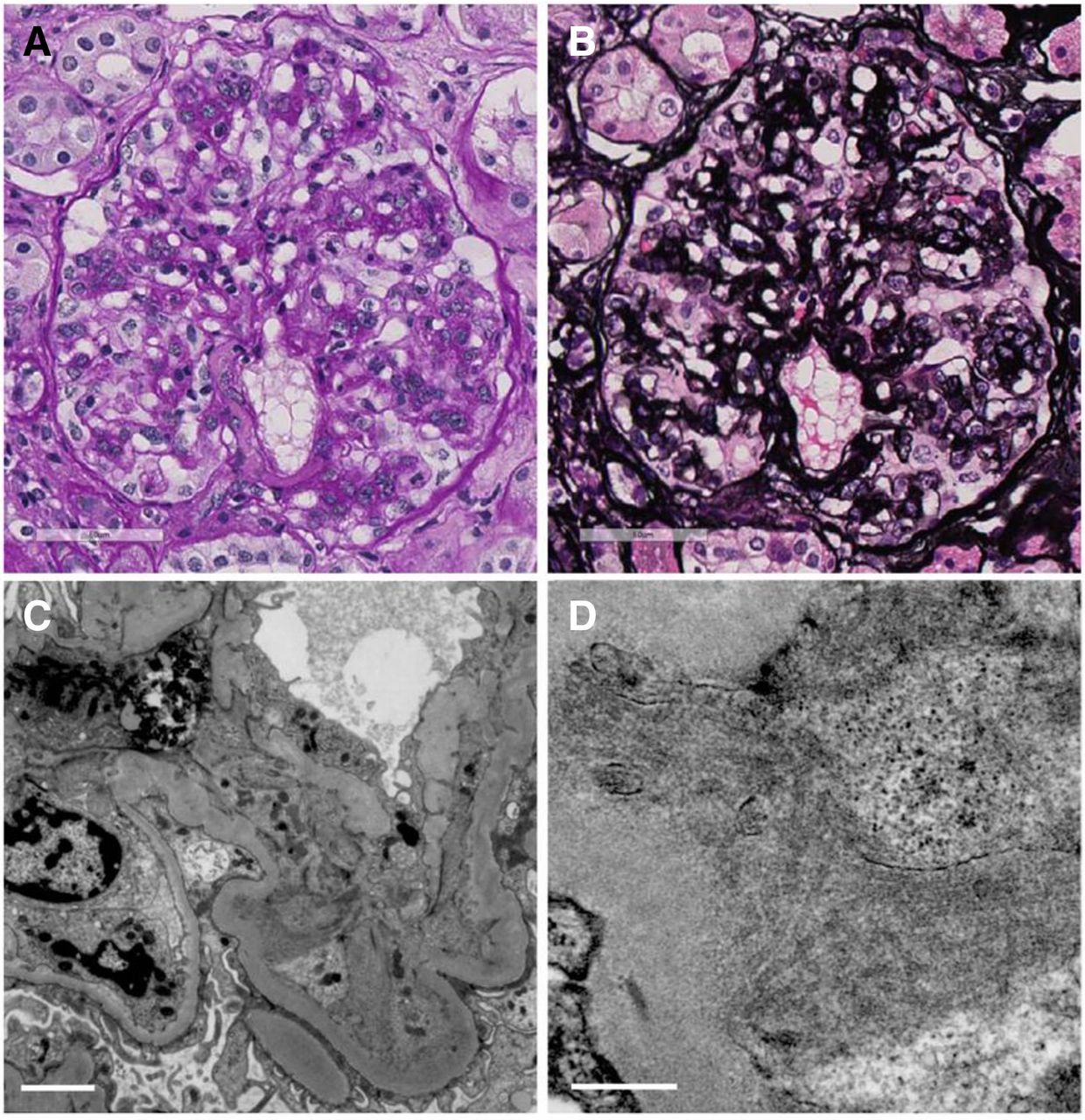
However, 2 years later, the patient’s proteinuria had increased to 6–9 g/gCr and her renal function had started to decline. In April 2017, at the age of 77, her serum creatinine level had increased to 1.07 mg/dL (eGFR 38 mL/min). Immunological screening was again unremarkable, except for a low serum C3 level of 65 mg/dL. Protein immunoelectrophoresis of serum and urine again did not show paraprotein. Serum-free kappa/lambda light chain ratio was within normal reference ranges (0.92). She underwent a second renal biopsy, which revealed 39 glomeruli, of which 16 were globally sclerosed. Twenty non-sclerotic glomeruli showed membranoproliferative glomerulonephritis (MPGN) with lobular accentuation, and endocapillary proliferative change was also noted in several glomeruli (figure 2A,B). Immunofluorescence studies revealed dominant C1q positivity on the capillary and reduced intensity in the mesangial areas. IgM and C3 were also positive in the capillary, while IgG, IgA and C4 were faintly positive in the capillary in a segmental manner (figure 3). Electron microscopy analysis showed dense deposits in the mesangial areas and subendothelial space. Higher magnification revealed hollow-centred microtubular structures of 35–45 nm diameter in these deposits, confirming the diagnosis of ITG (figure 2C,D). In addition, Congo-red staining of glomeruli under polarised light was negative, which excluded the possibility of amyloid fibrils.
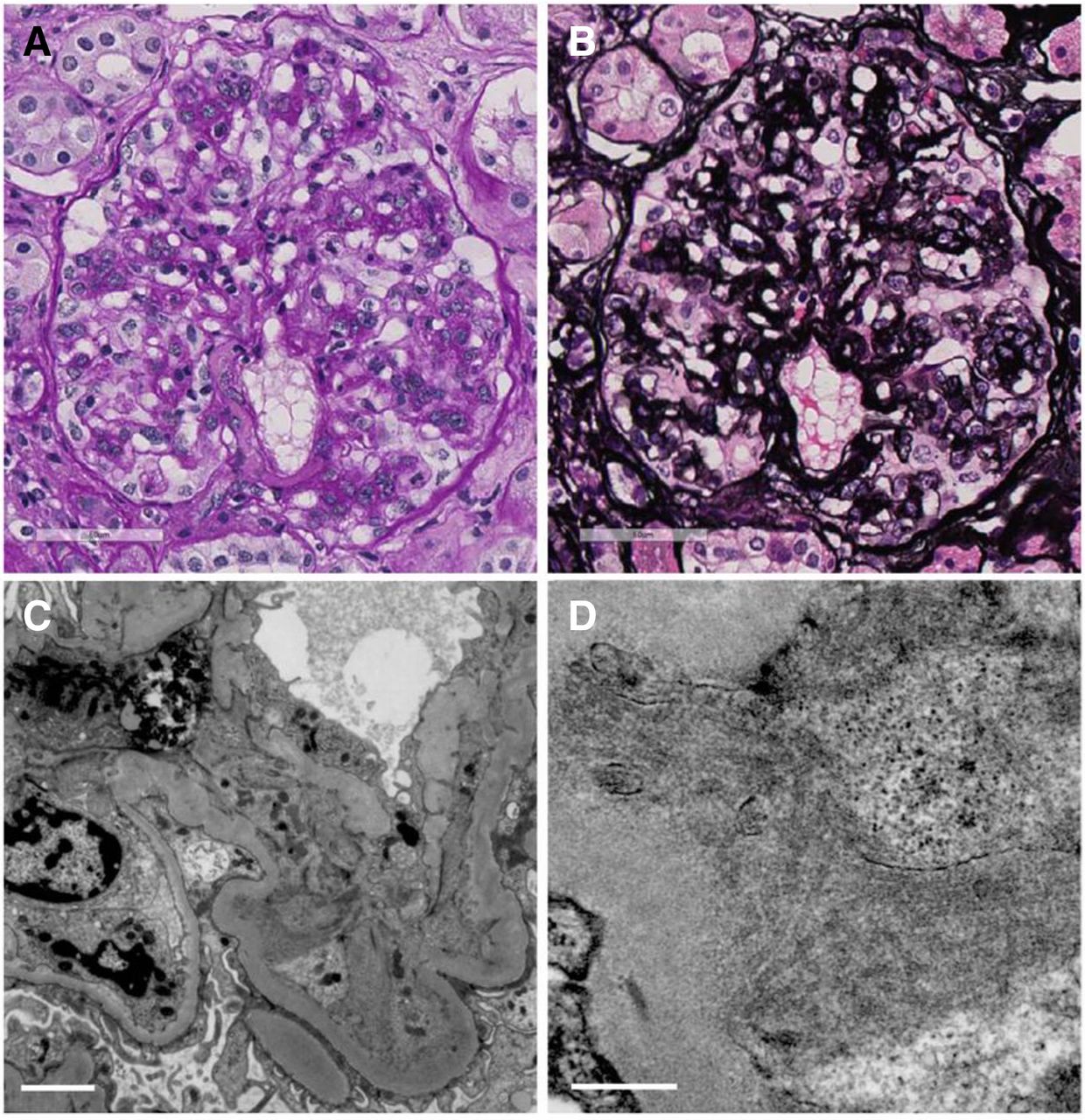
(A,B) Light microscopic findings from the second biopsy showed membranoproliferative glomerulonephritis with lobular accentuation, and endocapillary proliferative change. (A) Periodic acid-Schiff. 400×. (B) Periodic acid-Schiff-methenamine silver. 400×. (C,D) Electron microscopic findings showed dense deposits in the mesangial areas and subendothelial space. Higher magnification revealed hollow-centred microtubular structures of 35–45 nm diameter in these deposits. (C) The white line corresponds to 2 µm. (D) The white line corresponds to 500 nm.

Immunofluorescence findings from the second biopsy showed dominant C1q positivity on the capillary and reduced intensity in the mesangial areas. IgM and C3 were also positive in the capillary, while IgG, IgA and C4 were faintly positive in the capillary in a segmental manner.
Differential diagnosis
ITG belongs to the class of glomerular diseases characterised by the deposition of organised often fibrillar structures. These are divided into two categories consisting of Congo-red positive diseases, encompassing all types of amyloidosis, and Congo-red negative disorders including ITG, fibrillar glomerulopathy, cryoglobulinaemia, systemic lupus erythematosus and monoclonal immunoglobulin deposition disease.4 In the present case, Congo-red staining was negative, so we should think of the latter cases. These disorders can be distinguished on the basis of features of electron dense deposits. If the deposits are organised microfibrills like the present case, possible diagnoses are ITG, fibrillar glomerulopathy and cryoglobulinic glomerulopathy. In fibrillar glomerulopathy cases, deposits are randomly oriented and typically 16–24 nm in diameter, whereas, in ITG cases, deposits have distinct hollow cores and range from 10 to 90 nm in diameter, which corresponds well with the present case.5 Cryoglobulinic glomerulopathy typically has paired, curved 25–30 nm deposits, which were not seen in the present case. Besides, we did not observe lesions typical of cryoglobulinic nephropathy, macrophagic infiltrates and organised microtubular osmiophilic material in capillary lumens, or crystal inclusions within endothelial cells and macrophages.5 Therefore, ITG is the most compatible diagnosis.
Treatment
Treatment with oral prednisolone (30 mg/day; 0.8 mg/kg/day) was initiated, and the dosage was tapered by 5 mg every 2 weeks.
Outcome and follow-up
Proteinuria gradually decreased and complete remission was obtained 36 days after the initiation of immunosuppressive therapy (proteinuria 6.02 g/gCr (day 1) to 0.12 g/gCr (day 36)) (figure 4). The patient’s renal function stabilised after treatment, ranging between Cre 1.0 and 1.2 mg/dL.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes over time in uTP/Cre and eGFR after the initiation of treatment. On day 36, complete remission of proteinuria was obtained (uTP/Cr=0.12 g/gCr). eGFR, estimated glomerular filtration rate.
Discussion
ITG was first introduced by Schwartz and Lewis to describe a glomerular disease characterised by Congo-red negative microtubular deposits in the glomerulus.6 When cryoglobulinaemia and systemic lupus erythematosus are excluded, ITG is a very rare glomerular disease encountered in 0.06% of native kidney biopsies.7
What is notable for the present case is that renal biopsy was performed twice at an interval of 4 years. ITG is known to present in several pathological forms, although the natural course of pathological disease progression has not previously been clarified. In the present case, the first biopsy showed mesangial proliferation and focal moderate cellular infiltration of interstitium. On the second biopsy, severe endocapillary proliferation was added to mesangial proliferation, and double contouring of the basement membrane with mesangial interposition and lobulation of glomerulus was also revealed. These pathological changes indicated that ITG is a rapidly progressive disease featuring deposit expansion and endocapillary proliferation.
The pathological features of 16 ITG cases have previously been reported as shown in table 1.8
Pathological forms of 16 immunotactoid glomerulopathy cases
A correlation between histologic pattern, the severity of renal insufficiency and amount of proteinuria at presentation has previously been reported.7 Where ITG cases were too few to permit the analysis of correlations between histologic findings and the presentation of clinical features, the study reported the clinicopathological relationship with fibrillar glomerular nephritis (FGN). In FGN, the time to ESRD varied depending on histology under light microscopy. The median times were as shown in table 2.8
Median times to ESRD in fibrillar glomerulopathy cases
In the present case, the pathological change over time corresponds to the degree of severity described in this report.
Unfortunately, no successful controlled trials have been conducted to provide guidance for the appropriate therapy in patients with ITG, and no treatment to date has been proven to be effective. The clinical course in patients with ITG is characterised by renal insufficiency progressing to ESRD over 2–4 years in 50% of patients.1 9
In a study of 16 patients treated with prednisolone and/or cytotoxic agents, only 2 patients experienced partial remission of proteinuria, with the remaining 14 patients having no response.10 Another study reported that 10 out of 12 patients with ITG received one or more immunomodulatory therapy, of whom 4 patients had partial remission and 1 had complete remission.8 Overall, response to immunosuppressive therapy is reportedly poor.
The pathogenesis of ITG has not been fully clarified. Recent studies in CD2-associated protein (CD2ap) knockout mice provide support for a defect in glomerular function in ITG that is responsible for tactoid formation, indicating that the defect is localised to the podocyte.11 12 CD2ap-deficient mice have congenital nephrotic syndrome and compromised immune function13 as well as have mesangial and subendothelial deposits that comprise parallel arrays of microtubules similar to those seen in ITG.14 Therefore, it may be postulated that glomerular deposits in ITG are secondary to acquired defects in critical podocyte cellular functions involved in the clearance of filtered and retained immunoglobulin.2 If that is to be considered, we should clearly distinguish between idiopathic and secondary ITG, as the former originates from podocyte dysfunction whereas the latter arises from the production of restricted free light chains. Responsiveness to therapy can also differ between the disease forms, being generally poor in idiopathic ITG while secondary ITG with underlying haematological disease reportedly responds well to chemotherapy. In the Bridoux et al study, 10 of 12 nephrotic ITG patients with underlying lymphoproliferative disease and/or paraproteinaemia showed complete or partial remission of nephrotic syndrome after chemotherapy.3 Among idiopathic cases, a complete or partial response to treatment using steroids alone was seen in only two cases.8 14 One case underwent bone marrow biopsy, which revealed 5%–10%λ-restricted plasma cells, while the other case did not undergo bone marrow biopsy. Both cases presented with an MPGN pattern histologically, with dense deposits at the subendothelial space and mesangial area.
In several cases of ITG, haematological conditions were not significant, although bone marrow biopsy revealed the proliferation of free light-chain restricted plasma cells. In such cases, lymphocyte crystal-like inclusions containing immunoglobulin with the same subclass and light chain type as those of glomerular deposits are detected. Cases of ITG with such microtubular monoclonal immunoglobulin deposits are associated with overt or latent lymphoproliferative disease and may be improved by chemotherapy or immunosuppressive agents.3
Bone marrow biopsy can reveal the latent lymphoproliferative diseases. However, there might be a concern for overscreening, considering the pretest probability and its invasiveness. Bridoux et al suggested that immunoblotting is a useful way to detect paraproteinaemia not detected by immunoelectrophoresis alone. They reported that they could diagnose latent B-cell lymphoma, using that method.3
We are not able to determine whether the high responsiveness to the therapy in the present case can be explained by a latent lymphoproliferative disease since the patient did not undergo bone marrow biopsy or immunohistological staining for free light chain. However, we suggest, in such cases as showing high responsiveness to the therapy and glomerular depositions of monoclonal IgG, a strict monitor for haematological conditions and examinations such as Interleukin-2 receptor level, CT scan and, if possible, bone marrow biopsy are recommended.
Further accumulation of cases reporting high responsiveness to treatment is needed to clarify the specific factor that indicates high susceptibility to treatment.
Learning points
Immunotactoid glomerulopathy (ITG) is a glomerular disease characterised by Congo-red negative microtubular deposits in the glomerulus, which normally shows low responsiveness to any immunosuppressive therapies.
The progression of ITG is characterised by the deposit enhancement, endocapillary proliferation and global sclerosis. These histological changes correspond to renal survival.
There might be a specific subgroup which shows high responsiveness to immunosuppressive therapies. This subgroup might be characterised by latent lymphoproliferative diseases, which could be proved by a bone marrow biopsy.
Acknowledgments
The authors would like to acknowledge the following people for their contribution to the case study: Dr Eriko Kanehisa, Dr Yuta Nakano and Dr Megumi Yamamuro for their cooperation in medical practice.
References
Footnotes
Contributors AO wrote the manuscript with support from HF. JK and KN gave advice about pathological interpretation.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.